Materials
Unmodified DNA oligonucleotides, as well as DNA oligonucleotides modified with C3-azide and Cy3B, were purchased from MWG Eurofins and Metabion. The M13mp18 and p7560 scaffold was obtained from Tilibit. Magnesium chloride (1 M, no. AM9530G), sodium chloride (5 M, no. AM9759), ultrapure water (no. 10977-035), Tris (1 M, pH 8.0, no. AM9855G), EDTA (0.5 M, pH 8.0, no. AM9260G) and 10× PBS (no. 70011051) were purchased from Thermo Fisher Scientific. BSA (no. A4503-10G) was ordered from Sigma-Aldrich. Triton X-100 (no. 6683.1) was purchased from Carl Roth. Sodium hydroxide (no. 31627.290) was purchased from VWR. Paraformaldehyde (no. 15710) and glutaraldehyde (no. 16220) were obtained from Electron Microscopy Sciences. Tween-20 (no. P9416-50ML), glycerol (no. 65516-500 ml), methanol (no. 32213-2.5L), protocatechuate 3,4-dioxygenase pseudomonas (PCD, no. P8279), 3,4-dihydroxybenzoic acid (PCA, no. 37580-25G-F) and (±)-6-hydroxy-2,5,7,8-tetra-methylchromane-2-carboxylic acid (trolox, no. 238813-5G) were ordered from Sigma-Aldrich. Neutravidin (no. 31000) was purchased from Thermo Fisher Scientific. Biotin-labelled BSA (no. A8549) and sodium azide (no.769320) were obtained from Sigma-Aldrich. Coverslips (no. 0107032) and glass slides (no. 10756991) were purchased from Marienfeld and Thermo Fisher Scientific, respectively. Fetal bovine serum (FBS, no. 10500-064), 1× PBS (pH 7.2, no. 20012-019), 0.05% trypsin-EDTA (no. 25300-054), salmon sperm DNA (no. 15632011), OptiMEM (no. 31985062) and Lipofectamine LTX (no. A12621) were purchased from Thermo Fisher Scientific. Gold nanoparticles (90 nm, no. G-90-100) were ordered from Cytodiagnostics. Nanobodies against GFP (clone 1H1) with a single ectopic cysteine at the C terminus for site-specific conjugation were purchased from Nanotag Biotechnologies. DBCO-PEG4-Maleimide (no. CLK-A108P) was purchased from Jena Bioscience.
Buffers
The following buffers were used for sample preparation and imaging.
Buffer A: 10 mM Tris pH 8.0, 100 mM NaCl and 0.05% Tween-20
Buffer B: 10 mM MgCl 2 , 5 mM Tris-HCl pH 8.0, 1 mM EDTA and 0.05% Tween-20 pH 8.0
Buffer C: 1× PBS, 1 mM EDTA, 500 mM NaCl pH 7.4, 0.02% Tween, optionally supplemented with 1× trolox, 1× PCA and 1× PCD
Blocking buffer: 1× PBS, 1 mM EDTA, 0.02% Tween-20, 0.05% NaN 3 , 2% BSA, 0.05 mg ml –1 sheared salmon sperm DNA
Two-dimensional (2D) DNA origami folding buffer: 10 mM Tris, 1 mM EDTA, 12.5 mM MgCl 2 pH 8.0
3D DNA origami folding buffer: 5 mM Tris, 1 mM EDTA, 5 mM NaCl, 20 mM MgCl2 pH 8.0
1× TA buffer: 40 mM Tris pH 8.0, 20 mM acetic acid
PCA, PCD and trolox
Trolox (100×) was made by the addition of 100 mg of trolox to 430 μl of 100% methanol and 345 μl of 1 M NaOH in 3.2 ml of water. PCA (40×) was made by mixing 154 mg of PCA in 10 ml of water and NaOH and adjustment of pH to 9.0. PCD (100×) was made by the addition of 9.3 mg of PCD to 13.3 ml of buffer (100 mM Tris-HCl pH 8.0, 50 mM KCl, 1 mM EDTA, 50% glycerol).
DNA-PAINT docking and imager sequences
Four orthogonal DNA sequence motifs were used to label targets in four RESI rounds. The docking strands were 5xR1 (TCCTCCTCCTCCTCCTCCT), 5xR2 (ACCACCACCACCACCACCA), 7xR3 (CTCTCTCTCTCTCTCTCTC) and 7xR4 (ACACACACACACACACACA). The respective imagers were R1 (AGGAGGA-Cy3B), R2 (TGGTGGT-Cy3B), R3 (GAGAGAG-Cy3B) and R4 (TGTGTGT-Cy3B). The design of 2D RESI origami required extension of the R1 site at the 5′ end such that the adjacent R1 and R3 docking strands could be spaced apart by a single base pair. Thus, the docking strand 5′ 5xR1 (TCCTCCTCCTCCTCCTCCT) and the 5′ R1 imager (Cy3B-AGGAGGA) were used rather than the 3′ versions for both 2D DNA origamis.
DNA origami self-assembly (2D)
All 2D DNA origami structures were designed in caDNAno40. Self-assembly of DNA origami was accomplished in a one-pot reaction mix with a total volume of 40 μl, consisting of 10 nM scaffold strand (for sequence, see Supplementary Data 2), 100 nM folding staples (Supplementary Data 1), 500 nM biotinylated staples (Supplementary Data 1) and 1 μM staple strands with docking site extensions (Supplementary Data 1) in 2D DNA origami folding buffer. The reaction mix was then subjected to a thermal annealing ramp using a thermocycler. First, it was incubated at 80 °C for 5 min, cooled using a temperature gradient from 60 to 4 °C in steps of 1 °C per 3.21 min and finally held at 4 °C.
DNA origami self-assembly (3D)
The 3D DNA origami disk structure was designed in caDNAno40. Self-assembly of the DNA origami disk was accomplished in a one-pot reaction mix of 50 µl total volume, consisting of 20 nM scaffold strand p7560 (for sequence, see Supplementary Data 3), 200 nM core folding staples (Supplementary Data 1), 200 nM staple sequences without handle extension (Supplementary Data 1), 500 nM biotinylated staples (Supplementary Data 1), 2 μM staple strands with R4 docking site extensions and 4 μM staple strands with R1 or R3 docking site extensions (Supplementary Data 1) in 3D DNA origami folding buffer. The reaction mix was then subjected to a thermal annealing ramp using a thermocycler. It was first incubated at 80 °C for 5 min then cooled using a temperature gradient from 60 °C to 20 °C in steps of 1 °C h–1 and finally held at 20 °C.
DNA origami purification
After self-assembly, structures were purified by agarose gel electrophoresis (1.5% agarose, 1× TA, 10 mM MgCl 2 , 0.5× SybrSafe) at 4.5 V cm–1 for 1.5 h. Gel bands were cut, crushed and the origami stored in low-binding Eppendorf tubes at −20 °C.
DNA origami sample preparation and imaging
For sample preparation, a bottomless six-channel slide (ibidi, no. 80608) was attached to a coverslip. First, 80 μl of biotin-labelled BSA (1 mg ml–1, dissolved in buffer A) was flushed into the chamber and incubated for 5 min. The chamber was then washed with 360 μl of buffer A. A volume of 100 μl of neutravidin (0.1 mg ml–1, dissolved in buffer A) was then flushed into the chamber and allowed to bind for 5 min. After washing with 180 μl of buffer A and subsequently with 360 μl of buffer B, 80 μl of biotin-labelled DNA structures (approximately 200 pM) in buffer B was flushed into the chamber and incubated for 5 min. For measurement of the DNA origami disk, additional 2D DNA origami structures with 12 target sites9 spaced 20 nm apart were incubated together, with the 3D disk origami serving as fiducials for drift correction. After DNA origami incubation the chamber was washed with 540 μl of buffer B. For DNA origami disk structures, 150 μl of gold nanoparticles (diluted 1:10 in buffer B) was flushed through and incubated for 5 min before washing with 540 μl of buffer B. Finally, 180 μl of the imager solution in buffer B was flushed into the chamber. The chamber remained filled with imager solution and imaging was then performed. Between imaging rounds, the sample was washed three times with 1 ml of buffer B until no residual signal from the previous imager solution was detected. Then, the next imager solution was introduced. For RESI, two imaging rounds were performed with imagers R1 and R4 present in round 1 and the imagers R3 and R4 in round 2 (R1 and R3 probe the sites of interest for RESI and R4 serves alignment purposes).
Nanobody–DNA conjugation
Nanobodies were conjugated as described previously32. Unconjugated nanobodies were thawed on ice, then 20-fold molar excess of bifunctional DBCO-PEG4-Maleimide linker was added and reacted for 2 h on ice. Unreacted linker was removed by buffer exchange to PBS using Amicon centrifugal filters (10,000 MWCO). The DBCO-modified nanobodies were reacted with 5× molar excess of azide-functionalized DNA (R1, R2, R3 and R4) overnight at 4 °C. Unconjugated protein and free DNA were removed by anion exchange chromatography using an ÄKTA pure system equipped with a Resource Q 1 ml column.
Cell culture
CHO cells (CCL-61, ATCC) were cultured in Gibco Ham’s F-12K (Kaighn’s) medium supplemented with 10% FBS (no. 11573397, Gibco). U2OS-CRISPR-Nup96-mEGFP cells (a gift from the Ries and Ellenberg laboratories) were cultured in McCoy’s 5A medium (Thermo Fisher Scientific, no. 16600082) supplemented with 10% FBS. Cells were passaged every 2–3 days using trypsin-EDTA.
Nup96 EGFP imaging
U2OS-CRISPR-Nup96-mEGFP cells were seeded on ibidi eight-well high glass-bottom chambers (no. 80807) at a density of 30,000 cm–2. Cells were fixed with 2.4% paraformaldehyde in PBS for 30 min at room temperature. After fixation, cells were washed three times with PBS. Gold nanoparticles (200 μl) were incubated for 5 min and washed three times with PBS. Blocking and permeabilization were performed with 0.25% Triton X-100 in blocking buffer for 90 min. After washing with PBS, cells were incubated with 100 nM anti-GFP nanobodies in blocking buffer for 60 min at room temperature. To enable RESI, the nanobody solution consisted of 25 nM R1, R2, R3 and R4 docking-strand-coupled anti-GFP nanobodies with a total nanobody concentration of 100 nM. Unbound nanobodies were removed by washing three times with PBS, followed by washing once with buffer C for 10 min. Postfixation was performed with 2.4% paraformaldehyde in PBS for 15 min. After washing 3× with PBS, the imager solution in buffer C was flushed into the chamber. Between imaging rounds the sample was washed with 1–2 ml of PBS until no residual signal from the previous imager solution was detected. Then, the next imager solution was introduced. First, imagers R1, R2, R3 and R4 were added simultaneously to the sample to perform a standard DNA-PAINT measurement; then, RESI imaging was conducted via four subsequent imaging rounds with only one of the imagers.
Cloning
mEGFP-Alfa-CD20 was cloned by insertion of Alfa-CD20 into the mEGFP-C1 plasmid (no. 54759, Addgene). An Alfa-CD20 gblock (obtained from Integrated DNA Technologies) was amplified with primers cggcatggacgagct and gtacaagtccgga and, after cutting with restriction enzymes BsrGI and BamHI, Gibson assembly was performed (2× mix, NEB).
mEGFP-CD20 imaging
CHO cells were seeded on ibidi eight-well high glass-bottom chambers (no. 80807) the day before transfection at a density of 15,000 cm–2. Transfection with mEGFP-CD20 was carried out with Lipofectamine LTX as specified by the manufacturer. CHO cells were allowed to express mEGFP-CD20 for 16–24 h. Then, the medium was replaced with fresh F-12K medium + 10% FBS (in the untreated case) or with F-12K medium + 10% FBS + 10 ug ml–1 RTX-Alexa 647 (a gift from Roche Glycart) (in the RTX-treated case), followed by incubation for 30 min. After washing two times with fresh medium for 5 min, cells were fixed with 250 µl of prewarmed 4% PFA + 0.1% glutaraldehyde in PBS for 15 min. CHO cells were washed three times with PBS and quenched with 100 mM Tris pH 8.0 for 5 min. Permeabilization was carried out for 5 min with 0.2% Triton X-100 in PBS, followed by three washes with PBS. Cells were blocked in blocking buffer for 1 h at room temperature (RT). Anti-GFP nanobodies were incubated at a total concentration of 25 nM overnight at 4 °C; for RESI with four rounds this yielded 6.25 nM each of GFP-Nb-R1/2/3/4. After washing three times with PBS at RT for 15 min, cells were postfixed with 4% PFA at RT for 10 min followed by washing and postfixation as described above. Gold nanoparticles (90 nm) were diluted 1:3 in PBS and incubated for 10 min at RT and the sample was washed two times with PBS to remove unbound gold. The imager solution in buffer C for the first round was incubated for 5 min and then replaced with fresh imager, after which the first acquisition round was started. Between imaging rounds the sample was washed with at least 2 ml of PBS until no residual signal from the previous imager solution was detected. Then, the next imager solution was introduced. RESI imaging was conducted via four subsequent imaging rounds with only one of the imagers. In the final imaging round, imagers R1, R2, R3 and R4 were added simultaneously to the sample to perform a standard DNA-PAINT measurement.
Microscopy setup
Fluorescence imaging was carried out using an inverted microscope (Nikon Instruments, Eclipse Ti2) with the Perfect Focus System, applying an objective-type TIRF configuration equipped with an oil-immersion objective (Nikon Instruments, Apo SR TIRF ×100/numerical aperture 1.49, oil). A 560 nm laser (MPB Communications, 1 W) was used for excitation. The laser beam was passed through a cleanup filter (Chroma Technology, no. ZET561/10) and coupled to the microscope objective using a beam splitter (Chroma Technology, no. ZT561rdc). Fluorescence was spectrally filtered with an emission filter (Chroma Technology, nos. ET600/50m and ET575lp) and imaged on an sCMOS camera (Andor, Zyla 4.2 Plus) without further magnification, resulting in an effective pixel size of 130 nm (after 2 × 2 binning). The readout rate was set to 200 MHz. Images were acquired by choosing a region of interest of size 512 × 512 pixels. 3D imaging was performed using a cylindrical lens (Nikon Instruments, N-STORM) in the detection path. Raw microscopy data were acquired using μManager41 (v.2.0.1). Total internal reflection illumination was used for 2D and 3D DNA origami data, as well as for CD20 acquisition. Highly inclined and laminated optical sheet (HILO) illumination was employed for the acquisition of NPC data. Detailed imaging conditions for the respective experiments are shown in Extended Data Table 1.
Imaging parameters and duration
Due to target and sample heterogeneity the optimal imager concentration, c, used to achieve sparse blinking varies. Here we used concentrations from 100 pM (Nup96) to 800 pM (DNA origami). Optimal imager concentrations were determined visually for each sample. Concentrations were altered until blinking was frequent but sufficiently sparse to achieve good DNA-PAINT resolution.
The average number of expected binding events per binding site during a DNA-PAINT measurement is given by the duration of the measurement t measurement and the mean dark time τ dark (defined as \({\tau }_{{\rm{dark}}}=\frac{1}{{k}_{{\rm{on}}}\times c}\), with k on being the on-rate of a given imager strand) as:
$${n}_{{\rm{b}}{\rm{i}}{\rm{n}}{\rm{d}}{\rm{i}}{\rm{n}}{\rm{g}}{\rm{e}}{\rm{v}}{\rm{e}}{\rm{n}}{\rm{t}}{\rm{s}}}=\left(\frac{{t}_{{\rm{m}}{\rm{e}}{\rm{a}}{\rm{s}}{\rm{u}}{\rm{r}}{\rm{e}}{\rm{m}}{\rm{e}}{\rm{n}}{\rm{t}}}}{{\tau }_{{\rm{d}}{\rm{a}}{\rm{r}}{\rm{k}}}}\right)={t}_{{\rm{m}}{\rm{e}}{\rm{a}}{\rm{s}}{\rm{u}}{\rm{r}}{\rm{e}}{\rm{m}}{\rm{e}}{\rm{n}}{\rm{t}}}\times {k}_{{\rm{o}}{\rm{n}}}\times c.$$
The average number of localizations per binding event is given by the mean bright time τ bright and camera exposure time t exposure as
$${n}_{{\rm{locs}}{\rm{per}}{\rm{binding}}{\rm{event}}}=\left(\frac{{\tau }_{{\rm{bright}}}}{{t}_{{\rm{exposure}}}}\right).$$
Therefore, the average number of localizations expected per binding site over the course of the measurement is
$${n}_{{\rm{loc}}}=\left(\frac{{t}_{{\rm{measurement}}}}{{\tau }_{{\rm{dark}}}}\right)\times \left(\frac{{\tau }_{{\rm{bright}}}}{{t}_{{\rm{exposure}}}}\right)={t}_{{\rm{measurement}}}\times {k}_{{\rm{on}}}\times c\times \left(\frac{{\tau }_{{\rm{bright}}}}{{t}_{{\rm{exposure}}}}\right).$$
It follows that the total acquisition time necessary to collect n loc localizations is, on average,
$${t}_{{\rm{m}}{\rm{e}}{\rm{a}}{\rm{s}}{\rm{u}}{\rm{r}}{\rm{e}}{\rm{m}}{\rm{e}}{\rm{n}}{\rm{t}}}=\frac{{t}_{{\rm{e}}{\rm{x}}{\rm{p}}{\rm{o}}{\rm{s}}{\rm{u}}{\rm{r}}{\rm{e}}}\times {n}_{{\rm{l}}{\rm{o}}{\rm{c}}}}{{\tau }_{{\rm{b}}{\rm{r}}{\rm{i}}{\rm{g}}{\rm{h}}{\rm{t}}}{\times k}_{{\rm{o}}{\rm{n}}}\times c}.$$
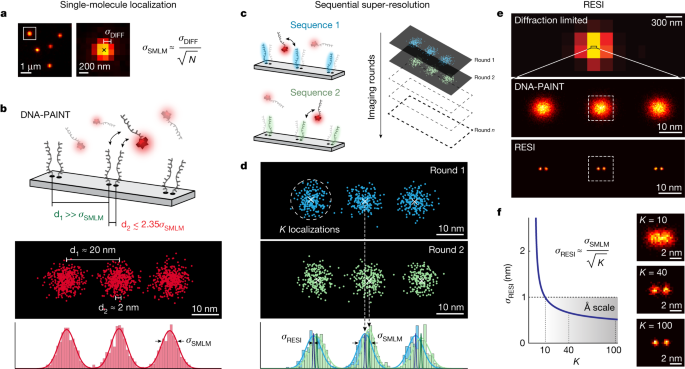
The necessary number of localizations, n loc , is calculated using \({\sigma }_{{\rm{RESI}}}=\frac{{\sigma }_{{\rm{DNA}} \mbox{-} {\rm{PAINT}}}}{\sqrt{{n}_{{\rm{loc}}}}}\), and thus \({n}_{{\rm{loc}}}={\left(\frac{{\sigma }_{{\rm{DNA}} \mbox{-} {\rm{PAINT}}}}{{\sigma }_{{\rm{RESI}}}}\right)}^{2}\) with the DNA-PAINT localization precision σ DNA-PAINT .
For expected imager concentrations between 50 and 800 pM, exposure times between 100 and 200 ms and kinetics reported previously32, the times required to collect 16 localizations (1 nm RESI precision given σ DNA-PAINT = 4 nm) vary between 42 s (R2, 800 pM, 100 ms exposure time) and 314 min (R5, 50 pM, 200 ms exposure time).
DNA-PAINT analysis
Raw fluorescence data were subjected to super-resolution reconstruction using the Picasso software package9 (latest version available at https://github.com/jungmannlab/picasso). Drift correction was performed with a redundant cross-correlation and gold particles as fiducials for cellular experiments, or with single DNA-PAINT docking sites as fiducials for origami experiments.
Channel alignment
Alignment of subsequent imaging rounds was performed iteratively in Picasso9, starting with a redundant cross-correlation and followed by gold fiducial alignment for cellular experiments. Every DNA origami was equipped with additional DNA-PAINT docking sites that were imaged simultaneously with the sites of interest in all imaging rounds, thus enabling their use as fiducials. First, redundant cross-correlation (2D and 3D origami measurements) and gold alignment (3D measurements) were performed in Picasso Render. To correct for nanoscopic movement of individual DNA origami during buffer exchange, channel alignment was not only performed on the full field of view but, additionally, small regions of interest containing only one DNA origami were selected. Within each region of interest, alignment was then conducted via the fiducial docking sites of the DNA origami. This was performed outside of Picasso in a custom Python script, not only to find the optimal translation between channels but also to correct for possible rotations of the DNA origami.
Clustering and RESI
Clustering of DNA-PAINT localizations
After channel alignment, DNA-PAINT data were analysed using a custom clustering algorithm for each imaging round. This algorithm is based on the fact that, in DNA-PAINT, localizations are independent measurements of the position of a target molecule and are observed to be Gaussian distributed. To assign localizations to a specific target molecule, we first used a gradient ascent method to find the centre of a localization cloud for each target. We then assigned all localizations circularly distributed around the centre point to the same target molecule. This is a valid approximation because, due to the reduction of effective target density by RESI’s sequential imaging approach, the majority of localization clouds from single targets are spaced sufficiently apart.
The clustering algorithm uses two input parameters: radius r, which sets the final size of the clusters and defines a circular environment around each localization, and the minimal number of localizations, n min , representing a lower threshold for the number of DNA-PAINT localizations in any cluster.
First, the number of neighbouring localizations within distance r from each localization is calculated. If a given localization has more neighbours within its r radius than all neighbouring localizations, it is considered a local maximum. If there are more than n min localizations within a circle of radius r around such a local maximum, these localizations are assigned to the same cluster; the remainder are not considered to be part of a cluster and are omitted from further analysis.
Further filtering of clusters is performed to exclude clusters that originate from unspecific sticking of imagers to the sample. Firstly, the mean frame (mean value of the frame numbers in which localizations occurred) of all localizations assigned to the same cluster is calculated. In the case of repetitive blinking the mean frame is expected to be around half the total number of frames42. The algorithm therefore excludes all clusters with a mean frame in the first or last 20% of frames. Secondly, sticking events in the middle of the acquisition time can be identified by dividing the acquisition time into 20 time windows each containing 5% of frames. If any of these time windows contains more than 80% of localizations in the cluster, it is excluded as a sticking event.
The choice of the clustering radius r and the threshold n min depend on the respective experimental conditions. A suitable value for n min can be estimated by picking localization clouds originating from single target molecules (that is, well separated) in Picasso Render, exporting pick properties and plotting a histogram of the number of localizations in each pick. n min is chosen to differentiate between populations corresponding to single targets and to background localizations.
The radius r scales with the size of the localization clouds and thus the localization precision. If too large a value is chosen, adjacent clusters might not be separated; if r is too small, ‘subclustering’ within one localization can occur. The latter also translates to a peak in NND at twice the clustering radius. A good a priori starting value for r is represented by approximately twofold the localization precision of the underlying DNA-PAINT measurement. Picasso Render offers a tool (Test Clusterer) in which the effect of different clustering parameters can be tested for a small region of interest.
For 3D clustering, an additional radius for the z direction is introduced because the spread of localizations in z is approximately twofold greater compared with x and y.
Calculation and rendering of RESI localization
Following cluster analysis, the centres of the DNA-PAINT localization groups were calculated as weighted (wtd) means by employing the squared inverse localization precisions \((\frac{1}{{{\text{lp}}}^{2}})\) as weights. For x and y coordinates:
$${\bar{x}}_{{\rm{w}}{\rm{t}}{\rm{d}}}=\frac{\mathop{\sum }\limits_{i=1}^{N}{w}_{i}{x}_{i}}{\mathop{\sum }\limits_{i=1}^{N}{w}_{i}},\,{w}_{i}=\frac{1}{{{\text{lp}}}^{2}}.$$
For z coordinates a standard mean without weights is used to calculate z positions. The precision of the resulting RESI localization is the weighted s.e.m. of the underlying grouped localizations:
$${({s}_{\bar{x}})}_{{\rm{wtd}}}=\frac{{({s}_{x})}_{{\rm{wtd}}}}{\sqrt{N}}=\sqrt{\frac{{\rm{Var}}{(x)}_{{\rm{wtd}}}}{N}},{\rm{where}}\,{\rm{Var}}{(x)}_{{\rm{wtd}}}=\frac{N}{N-1}\frac{{\sum }_{i=1}^{N}{w}_{i}{({x}_{i}-{\bar{x}}_{{\rm{wtd}}})}^{2}}{\mathop{\sum }\limits_{i=1}^{N}{w}_{i}}.$$
The choice for \(1/{{\text{lp}}}^{2}\) as weights is based on the following argument: under the hypothesis that localizations are independent and normally distributed with the same mean, the weighted mean based on inverse variances as weights is the maximum likelihood estimator of the mean of the whole set of localizations. Therefore, the variance of the weighted mean is minimal (the estimator is optimal) when the inverse variances of individual measurements \(1/{{\text{lp}}}^{2}\) are chosen as weights.
Finally, we take the average of the resulting x and y s.e.m. as the final precision of each RESI localization. For z coordinates the precision is estimated to be two times xy precision. Saving RESI localizations in a Picasso hdf5 file allowed us to render them as Gaussians with s.d. corresponding to their respective precision.
RESI resolution estimation
Evaluation of in silico RESI precision with numerical simulations
To evaluate the performance of RESI, in silico numerical simulations were performed. The algorithm consists of the following steps.
(1) A grid of defined positions of the binding sites (ground truth) is generated. Typically, a grid of positions was generated (Extended Data Fig. 1a, top left). (2) SMLM (DNA-PAINT) localizations are simulated as samples from a 2D Gaussian distribution with σ = σ SMLM . A large number (M) of localizations is generated per binding site (Extended Data Fig. 1a, top right). (3) For each binding site, subsets of K localizations are randomly selected (K << M). This results in \(n=\frac{M}{K}\) subsets of SMLM localizations (Extended Data Fig. 1a, bottom left) that are then averaged to generate n RESI localizations (Extended Data Fig. 1a, bottom right). (4) The resulting RESI localizations are then shown in a histogram (Extended Data Fig. 1b) and the trace (tr) of the covariance matrix is calculated. RESI precision is estimated as \({\sigma }_{{\rm{RESI}}}=\sqrt{\frac{1}{2}tr({\rm{cov}}\left(x,y\right))}\) (Extended Data Fig. 1c). This definition has been used before in the field as a scalar metric for 2D variance8. (5) Steps 3 and 4 are repeated for different values of K to numerically study σ = σ RESI (K).
Evaluation of experimental RESI precision by resampling of localizations
To evaluate the precision of RESI in experimental data, an analogous method was used. Briefly, the M total of DNA-PAINT localizations of each group corresponding to a single binding site was randomly resampled into subsets of K localizations, then steps 4 and 5 above were performed to evaluate σ RESI . The plotted σ RESI in Fig. 3d is the average value of all single binding sites in the dataset. Error bars represent the s.d. of the different σ RESI values calculated for different binding sites.
Note that this analysis can be performed only for K << M to have sufficient \(n=\frac{M}{K}\) RESI localizations for a statistically significant estimation. Because final RESI localization takes into account all M DNA-PAINT localizations, final precision is extrapolated as \({\sigma }_{{\rm{RESI}}}=\frac{{\sigma }_{{\rm{SMLM}}}}{\sqrt{M}}\).
Stochastic labelling: simulations and user guidelines
In RESI, the sparsity of binding sites in the sample is achieved by labelling a single species of biomolecules with different orthogonal DNA sequences. The labelling process is performed in a stochastic manner: n different labels (for example, DNA-conjugated nanobodies) targeting the same protein species are simultaneously incubated in the sample and thus the probability of each single protein being labelled with a certain sequence i (i = 1, …, n) is \({p}_{i}=\frac{1}{n}\), given that the same concentration of each label is used. Subsequently, n imaging rounds are performed to record all groups of localizations required to obtain the final RESI image.
The minimum number of labels (n) and rounds necessary to achieve sufficient sparsity of binding sites in each imaging round will depend mainly on three factors: SMLM localization precision and density and the molecular arrangement of the protein of interest. Here we describe how these parameters affect the final RESI results using a few practical examples.
Case 1: protein structure with oligomers not resolvable with DNA-PAINT
A typical study case is that of single proteins arranged in dimers, which in turn present another specific spatial organization in space. This is the case, for example, of the Nup96 in the NPC. In this case stochastic labelling has to be such that the probability of labelling two proteins forming a dimer with different sequences is sufficiently high. For n rounds of labelling/imaging, the probability is
$$P\left({\rm{diff}}\,.\,{\rm{seq}}.\right)=1-{p}_{i}=1-\frac{1}{n}$$
for n = 4 labelling/imaging rounds P(diff. seq.) ≈ 75%. We chose n = 4 to demonstrate that it provides a relatively high P(diff. seq.) with only a few imaging rounds. We note, however, that n > 4 could be used to increase P(diff. seq.) and hence to maximize the sparsity of labelled binding sites in each round.
To resolve a set of an arbitrary number of molecules, m, spaced more closely than the resolution of DNA-PAINT, they must be labelled with n orthogonal sequences. In general, the proportion of m molecules labelled with n orthogonal sequences, and thus the proportion of resolvable sets of molecules, follows the equation
$$P\left(m,n\right)=\frac{n!}{\left(n-m\,\right)!{n}^{m}}.$$
Case 2: proteins distributed similarly to CSR at a certain density
This is a common case—for example, for membrane receptors. If proteins are distributed in a CSR fashion (Extended Data Fig. 14a) at a given density, DNA-PAINT can already resolve single proteins that are sufficiently spaced from their NNs. We will consider that proteins at a distance d = 4 × σ DNA-PAINT are reliably resolved (note that this criterion is significantly stricter than 2.35 × σ DNA-PAINT ). Then, for a given density, the NND histogram can be computed and the fraction of distances below d calculated (Extended Data Fig. 14b). This represents the fraction of single proteins, F, that will not be resolved by DNA-PAINT. Here we plot F as a function of both density and resolution (Extended Data Fig. 14c). Such a map already provides a tool to understand the level of SMLM resolution needed to resolve single proteins at a given density.
RESI can be interpreted here as a way to reduce the effective density by splitting targets into different stochastically labelled subsets. Hence, the effective density of each round will be reduced according to the formula \(\rho =\frac{{\rm{density}}}{n}\). Extended Data Fig. 14d shows one-dimensional cuts of the 2D map to provide guidelines to choosing the number of orthogonal sequences (and hence imaging rounds) needed to be able to perform RESI efficiently. For example, for an initial resolution of 20 nm (σ = 5 nm), which is typical for DNA-PAINT in a cellular context, and a density of \({\rm{d}}{\rm{e}}{\rm{n}}{\rm{s}}{\rm{i}}{\rm{t}}{\rm{y}}=200\,\frac{{\rm{m}}{\rm{o}}{\rm{l}}{\rm{e}}{\rm{c}}{\rm{u}}{\rm{l}}{\rm{e}}{\rm{s}}}{\mu {{\rm{m}}}^{2}}\) (relatively high), n = 4 different sequences are sufficient to provide P(diff. seq.) ≈ 90% for proteins below d (Extended Data Fig. 14d). These proteins will then be resolvable by RESI.
Model-free averaging
Model-free averaging of Nup96 data was performed for both DNA-PAINT and RESI measurement of the same nucleus, as described by Wu et al.30. The respective Picasso hdf5 files were segmented in SMAP43 and saved in a file format compatible for averaging by employing plugins segmentNPC, NPCsegmentCleanup and sitenumbers2loc. Model-free averaging was then performed on the resulting _sml.mat files with default parameters by running the particleFusion.m script in Matlab (available with the SMAP source code). The averages shown correspond to the result of the final iteration, in which each point is rendered with a Gaussian of σ = 2 nm in x, y and z.
Numerical simulations for CD20 distribution
To interpret the results of the NND data in untreated cells, numerical simulations were performed. Briefly, two populations, one of CD20 monomers and one of dimers with a CSR distribution, were simulated and then their NNDs calculated. The algorithm can be summarized as follows:
(1) Choice of parameters. Density of monomers: number of monomers per unit area; density of dimers: number of dimers per unit area; dimer distance: expected distance between the two molecules including the labelling construct; uncertainty: variability in the position of each molecule due to labelling and localization errors; labelling efficiency: fraction of ground-truth molecules that will actually be labelled and measured. The observed density, which has to match the experimental parameter, then becomes observed density = (density of monomers + density of dimers) × labelling efficiency. For quantification of the labelling efficiency of the DNA-conjugated GFP nanobody we used a transiently transfected CHO cell line expressing a GFP- and Alfa-tag at the C terminus of a monomeric membrane protein (for example, CD86). We then labelled GFP- and Alfa-tag using their cognate nanobodies conjugated to two orthogonal docking sequences and performed two rounds of Exchange-PAINT. We then obtained the best-fitting parameters for a sample comprising pairs of GFP/Alfa-tag, and isolated Alfa-tags, similarly to how CD20 dimer/monomer analysis is performed. The ratio of these two populations is then used as an estimation of labelling efficiency. Full details of the quantification approach will be available in a manuscript currently in preparation. (2) Simulation of monomers: a set of spatial coordinates with CSR distribution and given density are drawn; simulation of dimers: a set of spatial coordinates with CSR distribution are drawn, representing the centre of each dimer. For each dimer centre, two positions are generated with a random orientation and a distance with expected value dimer distance. The position of each pair of molecules is drawn, taking into account the uncertainty parameter (drawn from a Gaussian distribution). (3) A random subset of ‘detectable’ molecules is taken from the ground-truth set (fraction = labelling efficiency) to simulate the labelling process. (4) NNDs are calculated on the subset of detectable molecules.
The parameters density of monomers = 212 μm–2, density of dimers = 0 μm–2, uncertainty = 5 nm and labelling efficiency = 50% were used to compare data for RTX-treated cells with a CSR distribution of monomers.
For the untreated case, the best-fit parameters were obtained through an iterative, nonlinear, least-squares algorithm. The experimentally observed density (50 molecules µm–2) is used for the simulation.
Description of the iterative nonlinear, least-squares algorithm
For every set of parameters a simulation is performed, NNDs are histogrammed and the sum of the squared differences between the simulation and experimental histogram are computed. A fit consists of finding the parameters that minimize the sum of the squared differences.
Parameters
D, average dimer distance (nm)
σ_label, variability introduced by the labelling (nm)
frac_of_dimers, fraction of dimers (%)
Note: frac_of_monomers = 100 – frac_of_dimers
Estimation of parameters
(1) Coarse-fit over a large range of parameters to determine the range of the best-fit parameters. Range D = 1–20 nm, σ_label = 1–20 nm, frac_of_dimers = 0–100%. (2) Fine-fit over a reduced parameter space around the best-fit results in the previous step.
The parameters D_opt, σ_label_opt and frac_of_dimers_opt that best match the proposed model and the data are now found. In this case it resulted in D_opt = 13.5 nm, σ_label_opt = 5.5 nm, frac_of_dimers_opt = 47% (Fig. 4e,f).
Estimation of parameter uncertainty
(1) M is created (in this case, M = 100), simulated (using datasets D_opt, σ_label_opt, frac_of_dimers_opt) with the same number of molecules as the experimental data (around 21,000). (2) M datasets are fine-fitted and the best-fit parameters D_opt, σ_label_opt and frac_of_dimers_opt are obtained. Three sets are obtained: D_opt, σ_label_opt and frac_of_dimers_opt. (3) The distributions of D_opt, σ_label_opt and frac_of_dimers_opt are studied. Standard deviation can be used as an estimation of the parameter uncertainties obtained in b.
The uncertainties of the parameters D_opt, σ_label_opt and frac_of_dimers_opt are now obtained.