Ethics statement
White-tailed deer were captured under a wildlife damage management agreement administered by USDA/APHIS Wildlife Services. Sample collection was conducted opportunistically in collaboration with regulatory agencies and as part of routine surveillance programs. Sex and gender were not excluded in this study. This study focuses on viral evolution and transmission and did not include an analysis of host gender and sex.
To compare intra-host intra-host single nucleotide variations between human and white-tailed deer SARS-CoV-2 viruses, the de-identified human samples testing positive for SARS-CoV-2 were selected from a cohort of routinely collected samples at University of Missouri Health Care which covers the Columbia, MO and the neighboring counties, and this study was approved by the University of Missouri Institutional Review Board (#2025449). Sex and gender were not excluded in this study. No targeted recruitment efforts were undertaken.
White-tailed deer sample collection
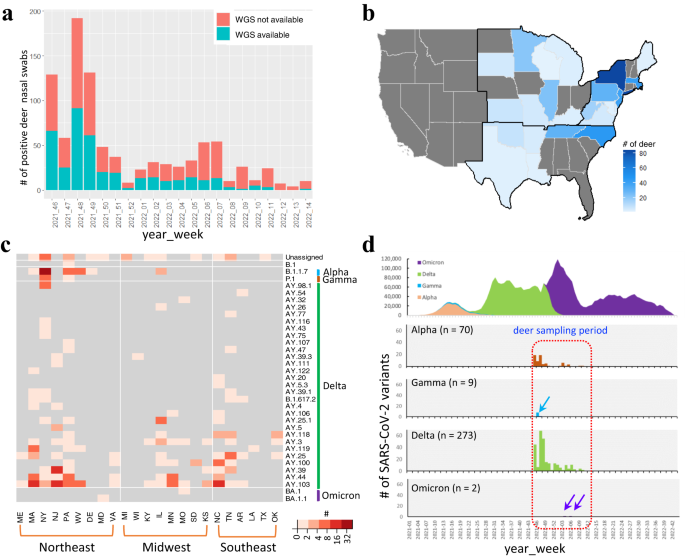
From November 4, 2021 to April 4, 2022, a total of 8830 white-tailed deer nasal swab samples were collected from Washington, D.C. and 26 participating states in the US either by hunter- or agency-harvest. None of the animals sampled exhibited clinical signs of SARS-CoV-2 infection. Over 95% of the white-tailed deer population resides in the Northeast, Midwest, and Southeastern United States, represented by Washington, D.C. and 26 participating states in the US from which the samples were collected46. These regions are also where much of the 6 million white-tailed deer are harvested annually by hunters47. In the vast majority of cases, a paired blood sample was collected onto a Nobuto filter strip for serological analysis.
RNA extraction and qRT-PCR
SARS-CoV-2 RNA was prepared from oral and nasal swab samples preserved in PrimeStore Molecular Transport Media (MTM, Longhorn Vaccines and Diagnostics LLC, catalog# LH105) using MagMAX™ CORE Nucleic Acid Purification Kits (Applied Biosystems, catalog# A32702) in accordance with the manufacturers’ instructions. 5 µL of RNA extract was for qRT-PCR detection of SARS-CoV-2 N1 and N2 targets using the BioRad Reliance One-Step Supermix Kit (catalog# 12010221) with SARS-CoV-2 RUO Primers & Probes obtained from Integrated DNA Technologies (catalog# 10006713). Reaction and thermocycling conditions were identical to those described for the BioRad Reliance SARS-CoV-2 RT-PCR assay, and data was acquired BioRad CFX96 Touch Real-Time PCR Detection System or CFX Opus Real-Time PCR System.
Genomic sequencing and assembly
For SARS-CoV-2 whole genome RT-PCR amplification, cDNA libraries were prepared using the Nextera XT DNA Sample Preparation Kit (catalog# FC-131-1096), and sequencing was performed using the 500 cycle MiSeq Reagent Kit v2 (catalog# MS-102-2003) according to manufacturer instructions48. The quality of paired-end reads obtained from MiSeq sequencing were analyzed and trimmed with a Phred quality score of 20, which indicates a base call accuracy of 99%, the likelihood of finding 1 incorrect base call among 100 bases49. The sequence assembly and consensus sequence construction were conducted with the Wuhan-Hu-1 (Accession Number: NC_045512.2) as the reference genome by using the Iterative Refinement Meta-Assembler (IRMA, v1.0.2)50. Manual curation and validation were performed for those genomic positions with low coverage. IRMA was used to analyze single nucleotide variants (SNVs) and assess intra-host viral genomic diversity. A minority iSNV was identified when the frequency of an allele was at least 5% but less than 50% among the reads. In addition, the consensus sequences were validated by using Qiagen CLC Genomics Workbench 20.0.4 with a quality score of 0.05.
Genomic sequence alignment and molecular characterization
NextStrain51 was used to align SARS-CoV-2 genomic sequences and also to identify the nucleotide and amino acid substitutions between the white-tailed deer SARS-CoV-2 sequences and the associated human precursor SARS-CoV-2 sequence. Because of low coverage at the 5’ untranslated region (before position 266) and 3’ untranslated region (after position 29,674) of the genome, we excluded these positions from nucleotide and amino acid substitution analyses.
PANGOLIN lineage classification
The Phylogenetic Assignment of Named Global Outbreak Lineages (PANGOLIN) software (v4.0.5)20 was used (PANGO v4.0.6 (2022-04-22)) to determine Pango lineages for each white-tailed deer sequence. Genetic lineage classification was achieved for 355 sequences. Among these samples, only those with a high sequencing coverage over >95% of the reference genome (n = 282), an IRMA score of 95%, were selected for further evolutionary analyses.
Bayesian phylogenetic analyses
Time-scaled Bayesian analyses were performed by using the Markov chain Monte Carlo (MCMC) method with Bayesian phylogenetic analysis by sampling trees (BEAST)52 v.1.10.4 software. Each tree was generated by a HKY substitution model with gamma = 4, and a coalescent exponential growth prior with strict clock. The MCMC chain length was set as 30 to 100 million iterations with subsampling every 10,000 iterations. The babette53 R package v.2.3.2 was used for batch BEAST processing. Tracer v1.7.2 was used to assess the results. The maximum clade credibility (MCC) tree was summarized by using TreeAnnotator54 v.1.10.4, with a burn-in rate of 20% and the estimated time of divergence being represented by the median node height. The ggtree55 R package v3.8.0 was used for tree visualization.
Phylogeographic analyses
To determine whether SARS-CoV-2 was transmitted among white-tailed deer populations within the same geographic area or across different geographic areas, we performed Bayesian phylogeographic analyses by using BEAST52 (v.1.10.4). The Bayesian Stochastic Search Variable Selection (BSSVS) analysis was performed with county as discrete traits for the white-tailed deer SARS-CoV-2 sequences. Transmission events were filtered for statistical significance using the criteria of Bayes factor56 and posterior probability >0.7, as well as geographically nearby counties. Different statistical support levels were defined as follows: 3 \(\le\) Bayes factor \(\le\) 10 indicates support; 10 \(\le\) Bayes factor \(\le\) 100 indicates strong support; 100 \(\le\) Bayes factor \(\le\) 1000 indicates very strong support; and Bayes factor \(\ge\) 1000 indicates decisive support.
Identify potential precursor virus for white-tailed deer SARS-CoV-2 viruses
To identify potential precursor virus for a white-tailed deer SARS-CoV-2 virus, we grouped all human and white-tailed deer SARS-CoV-2 genomes by state and Pango lineages. As a result, 89 datasets were obtained, each containing human and white-tailed deer SARS-CoV-2 sequences from the same state and the same Pango lineage. To ensure the robustness of our selection, two methods were used to select the closest sequences for each white-tailed deer SARS-CoV-2 sequence: 1) FastTree57 v1.4.4 was used to identify those sequences based on topology; 2) Complete Composition Vector (CCV) v1.0 method58 was used to identify those sequences based genetic distances, and CCV is an alignment free methods to enable genetic distance measurement at large scale. The unique sequences from top 20 ranked sequences from each method were identified (Supplementary Data 10) and used for Bayesian phylogenetic analyses. Biopython v1.79 package59 were used for sequences data processing. The following criteria were used to determine whether a human SARS-CoV-2 genome was a precursor virus for a testing white-tailed deer SARS-CoV genome: 1) the genomes shall belong to the same genetic cluster with posterior probability ≥0.70; 2) the nucleotide identity shall be at least 99.85%. To ensure our analyses will not rule out SARS-CoV-2 viruses that could be from those viruses circulating in white-tailed deer (e.g., reported by previous studies) and other animals, we performed similar analyses including viruses from non-human hosts. Results showed that the white-tailed deer SARS-CoV-2 sequences from this study are genetically diverse from those reported earlier (Fig. 10). In total, we identified a human precursor virus for 264 out of the 282 white-tailed deer SARS-CoV-2 sequences analyzed, while no human precursor virus from the same state was identified for the remaining 18 sequences.
Fig. 10: The maximum clade credibility tree illustrating the evolutionary relation between the white-tailed deer SARS-CoV-2 sequences collected from this study and those from public database. A total of 332 white-tailed deer SARS-CoV-2 genomes were downloaded from GISAID (Global Initiative on Sharing All Influenza Data) on December 5, 2022. Among these genomes, 118 had complete and high coverage sequences (>99% coverage) and were included in the evolutionary analyses along with the white-tailed deer SARS-CoV-2 samples collected in this study. The timescale of the phylogenetic tree was represented in units of years, and the scale bar indicates the divergence time in years. Full size image
It is possible that transmission could occur between white-tailed deer and out-of-state travelers, such as through hunting or contact with animals. To ensure inclusion of potential out-of-state transmission events, we used Ultrafast Sample placement on Existing tRees (UShER) on all 14.3 million public SARS-CoV-2 genomic data as of March 30, 2023. UShER implements the Fitch-Sankoff algorithm to infer the placement of mutations on a given tree and on the variant list and mutation-annotated tree60. For 264 white-tailed deer sequences with a matching human sequence from the same state in our above analyses, UShER identified genetically close virus sequences for 230 from the same state and 30 from out-of-state human sequences. For the 18 white-tailed deer sequences without any matching human sequences from the same state in our previous analyses, UShER identified genetically close sequence viruses for all of them.
The phylogenetic analysis of spillover events was performed using a combination of out-of-state human SARS-CoV-2 sequences obtained from UShER and within-state human sequences obtained from FastTree and CCV analyses. The sequences were selected based on the following criteria: (1) they belonged to the same Pango lineages as the matched white-tailed deer sequences; (2) they were collected only in the US; and (3) their collection date was either earlier than or the same as the collection date of the white-tailed deer samples.
Identify independent spillover events from humans and transmission events within the white-tailed deer population
To identify independent potential spillover events of SARS-CoV-2 from humans and understand the transmission dynamics of SARS-CoV-2 viruses in the white-tailed deer populations, three types of clusters were defined, Human-Deer, Human-Deer-Deer, and Human-Deer-Human. For all these clusters, at least one human precursor virus was identified; those without human precursor virus were excluded from this analysis. For 282 white-tailed deer SARS-CoV-2 sequences identified, we were able to assign 238 of them to one of these three clusters (Supplementary Data 2). The following specific criteria are used for each cluster.
Human-Deer: 1) the human precursor virus and a single white-tailed deer SARS-CoV-2 sequence; 2) all human and SARS-CoV-2 sequences were from the same state and belonged to the same Pango lineage; 3) the posterior probability (PP) > 0.7 for the human-deer branch; and 4) the nucleotide sequence identities between the human precursor sequence and the white-tailed deer SARS-CoV sequence was ≥99.85%.
Human-Deer-Deer: 1) the human precursor virus and at least two white-tailed deer SARS-CoV-2 sequences; 2) all human and SARS-CoV-2 sequences were from the same state and belonged to the same Pango lineage (single white-tailed deer SARS-CoV-2 sequence from a different state inside would be removed from this event); 3) the posterior probability (PP) > 0.7 for the human-deer branch; and 4) the nucleotide sequence identities between the human precursor SARS-CoV sequence and at least one of the white-tailed deer SARS-CoV sequences ≥99.85%.
Human-Deer-Human: 1) the human precursor virus, at least two white-tailed deer SARS-CoV sequences, and another human SARS-CoV-2 sequence; 2) all human and SARS-CoV-2 sequences were from the same state and belonged to the same Pango lineage; 3) the human2 and at least one of the white-tailed deer SARS-CoV-2 sequences formed a deer-human2 sub-branch; 4) PP > 0.7 for both human1-the deer branch and the deer-human2 subbranch; 4) the nucleotide sequence identity between human1 and at least one of white-tailed deer SARS-CoV-2, and that between human2 and at least one of white-tailed deer SARS-CoV-2 was ≥99.85%.
Positive and negative selection analyses
Positive selection occurs when the rate of nonsynonymous substitutions (β) is greater than the rate of synonymous substitutions (α), while negative selection occurs when β < α. Both positive and negative selection analyses were performed using the FUBAR (Fast, Unconstrained Bayesian AppRoximation), from the HyPhy software v2.5.42(MP)61, 62. The phylogenetic trees generated by FastTree of each gene, were used in the analyses with the white-tailed deer sequences marked to test positive or negative selection in a specific branch in the phylogenetic tree. We considered sites with a posterior probability >0.9 to be significant, indicating either positive selection (prob(α < β)) or negative selection (prob(α > β))61.
Identification of repeated amino acid substitutions in white-tailed deer sequences
To understand whether amino acid substitutions could occur independently after the virus was introduced to the white-tailed deer population, we attempted to identify those repeated amino acid substitutions across the clusters of Human-Deer, Human-Deer-Deer, and Human-Deer-Human. A repeated amino acid substitution was defined with the following criteria: 1) the substitution was observed at least twice across those 106 within-state clusters of Human-Deer, Human-Deer-Deer, and Human-Deer-Human; 2) the substitution was not shown in all human SARS-CoV-2 viruses in the same lineage from the same state; 3) the substitution had a low frequency (e.g., <0.15%, obtained by Outbreak.info R package v0.2.063) in all SARS-CoV-2 human viruses from the US and worldwide. If a repeated amino acid substitution was under positive selection, it was defined as a white-tailed deer-adaptive substitution.
Parameter optimization for evolutionary analyses
To improve the accuracy and reliability of our analyses and mitigate potential issues arising from specific genomic positions that are prone to sequencing errors and recombination, we explored a masking approach to refine our analyses. Specifically, we masked 265 problematic positions as reported (source: https://github.com/W-L/ProblematicSites_SARS-CoV2), followed by phylogenetic analyses. The resulting tree topologies derived from the complete genome tree (see above description) and the tree with the masked positions exhibited a striking similarity (Supplementary Data 12). By excluding the reported positions from the tree, we performed a reanalysis of the transmission events, and the number of transmission events remained unchanged when using the same identification rules. We also conducted an assessment and determined that none of the white-tailed deer-specific adaptive substitutions were found within the 265 problematic positions. Therefore, it appears that the masked positions do not significantly impact the analyses conducted in this study. Therefore, all analyses conducted in this study were carried out without applying a mask to the 265 positions mentioned.
The genomic sequences of the majority of the white-tailed deer SARS-CoV-2 viruses and their corresponding potential human SARS-CoV-2 precursor viruses obtained from public databases demonstrated a high genomic nucleotide sequence identity, surpassing 99.80% (Supplementary Fig. 2a). To assess the reliability of the parameters utilized in defining transmission events, namely sequence identity and posterior probability, we conducted additional analyses encompassing various parameter ranges. Illustrated in Supplementary Fig. 2b–e, our findings revealed that the number of spillover events remained quite consistent when employing a sequence identity cutoff of 99.85% or lower. For instance, we detected a total of 110 spillover events with a sequence identity of 99.80% or lower. However, as the sequence identity cutoff became more stringent, the number of clusters decreased notably due to the lack of corresponding human SARS-CoV-2 precursor viruses. Specifically, we identified 100 spillover events with a sequence identity of 99.90% and only 66 events with a sequence identity of 99.95%. Conversely, the posterior probability did not have a notable impact on the number of transmission events, as the tree subclades associated with the majority of these events exhibited a posterior probability of 0.90 or higher. Only the subclade associated with one human-Deer-human spillover event had a posterior probability of 0.78 and another one with human-Deer spillover event had a posterior probability of 0.66. These parameter tuning analyses further confirm the robustness of the criteria employed in the analysis of transmission events. Specifically, a sequence identity cutoff of 99.85% and a posterior probability of 0.70 have been validated as reliable and effective thresholds.
Serological assays
Antibodies were extracted from Nobuto filter paper strips (Sterlitech, catalog # 49010010) and screened at the United States Department of Agriculture, National Wildlife Research Center (Fort Collins, USA) at a functional dilution of 1:20. Extracted samples were screened using a surrogate virus neutralization test (sVNT, Genscript cPass™, catalog# L00847-A) with data acquired using a VarioSkan Flash or Varioskan LUX multimode microplate reader (Thermo Fisher)16. At least two technical replicates were used for the calculation of the average % inhibition. The sVNT has not been validated for deer; however, previous evaluations with white-tailed deer sera samples suggested that sVNT results were qualitatively similar to a highly specific SARS-CoV-2 virus neutralization test16.
Structural modeling
Structural templates of different functions with genes of the SARS-COV-2 were downloaded from the Protein Data Bank (PDB)64. White-tailed deer sequences were aligned with the template sequences by MUSCLE v5.165. Visualization was conducted by PyMOL v2.5.466.
Public datasets
All available SARS-CoV-2 genomic sequences (n = 11,778,398 by 2022/07/09) from humans were downloaded from GISAID, and additional genomes (n = 1,020,486 by 2022/04/07) were curated from GenBank. From these sequences, human SARS-CoV sequences were selected from the 23 states where we sampled the white-tailed deer sequences. After removing redundant entries and selecting the complete and high coverage sequences, a total of 717,717 SARS-CoV-2 genomic sequences were obtained for this study. In addition, on December 5, 2022, we downloaded a total of 332 white-tailed deer SARS-CoV-2 genomes from GISAID. Among these genomes, 118 had complete and high coverage sequences (>99% coverage) and were included in the evolutionary analyses along with the white-tailed deer SARS-CoV-2 samples collected in this study. All these publicly available sequences and associated metadata used in this dataset are published in GISAID’s EpiCoV database and NCBI SARS-CoV-2 Resources.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.