Manufacturing of collagen-based corneal stromal equivalent
We developed a collagen-based corneal stromal substitute, BPCDX. BPCDX was manufactured following principles of good manufacturing practices (GMP), in a Class 5 (according to ISO-14644-1) air quality laminar flow clean-room facility in Linköping, Sweden by LinkoCare Life Sciences AB. Although the BPCDX is subject to an end-stage sterilization process, substantial efforts are made throughout the manufacturing process to maintain raw materials, intermediate products and the final product as aseptic as possible.
In brief, the fabrication procedure was as follows. Medical-grade purified freeze-dried type I porcine dermal atelocollagen was purchased from SE Eng Company (South Korea). The collagen was dissolved in PBS at room temperature to form a 5% collagen solution. The collagen solution was then exposed to a controlled vacuum evaporation29, and crosslinkers 1-[3-(dimethylamino) propyl]-3-ethylcarbodiimide methiodide (EDCM), N-hydroxysuccinimide (NHS) and riboflavin (vitamin B2) (Care Group, Vadodara) were added at 1% (w/v) ratios. The solution was mixed thoroughly and dispensed into custom curved contact lens molds with spacers of 280 µm and 440 µm used to delineate the final device thickness. Molds were clamped and samples were cured at room temperature. Removal from molds was achieved by immersion in PBS for 1 h at room temperature. Finally, BPCDX was rinsed with sterile PBS to extract any reaction residues. Following this chemical crosslinking, a second photochemical crosslinking of samples was performed by exposure to ultraviolet A (UVA) light, following the protocol of UVA–riboflavin corneal collagen crosslinking commonly used clinically32. We employed this second photochemical crosslinking step aiming to further improve the BPCDX mechanical strength, resistance to degradation and long-term stability. Notably, the BPCDX differs from single-crosslinked recombinant human-collagen-based implants evaluated previously in humans27,28,62 in several important ways. Firstly, we used medical-grade and FDA-approved porcine dermal collagen as a starting material, the ultra-purity of which results in a more mechanically robust hydrogel and reproducible batch-to-batch mechanical properties. The use of a novel vacuum evaporation technique enables a high collagen content (12–18%) to be controllably achieved29, relative to previous human-tested implants limited to 10% collagen by weight (the native human cornea is 13.6% collagen)27. We additionally increased the ratio of chemical crosslinkers to collagen from 0.4:1 to 1:1 to achieve a higher degree of crosslinking in comparison to earlier implants. Further, our use of the crosslinker EDCM29 (as opposed to EDC27) and subsequent UVA–riboflavin crosslinking with optimized dose and exposure parameters, differentiates the present BPCDX from previous collagen-based hydrogels, imparting additional strength and resistance to degradation (see Supplementary Table 1 for full comparison of manufacturing parameters and material properties).
Packaging of BPCDX
After manufacture, BPCDX is extracted from any reaction residues and rinsed thoroughly with sterile phosphate-buffered saline (PBS) in class 5 (Class 1000) laminar flow hoods and stored in sterile PBS in a sterilized, sealed blister-packed container as shown in Supplementary Fig. 4a. Primary blister packs are labeled according to ISO 15223-1:2012 and EN 1041:2008 as shown in Supplementary Fig. 4b. The entire blister cup package and instructions for use are inserted into a small outer packaging box for protection during transportation and storage (Supplementary Fig. 4c).
Sterilization of BPCDX
To provide an additional level of device safety beyond an aseptic manufacturing process, we implemented an additional end-stage sterilization procedure. Conventional sterilization techniques (for example, dry heat, steam, ethylene oxide, gamma irradiation and electron beam irradiation) widely used for rigid medical device sterilization have not been tested or validated for soft, tissue-engineered devices such as hydrogels, where they are likely to result in denaturation and loss of device integrity. For this reason, we developed a sterilization procedure using a pulsed UVC irradiation system (Xenon Z-1000 Flash UV Lamp System). Pulsed UV light is known for its ability to inactivate microbes, is widely used in the food industry and is gaining popularity as a sterilization method63,64.
Sterilization validation of BPCDX implants was performed according to ISO 14937:2016 and ISO 11137-1-3:2017 sterilization standards. Two sets of product samples were exposed to pulsed UVC light at two sterilization doses separately, and then tested for key properties and sterility to investigate if the UVC exposure at these dosages impacted BPCDX properties and/or packaging. Sterility tests were both performed internally using tryptic soy broth sterility test method and externally by an ISO-certified microbiology lab (MIKROLAB Stockholm AB) for independent sterility and bioburden tests.
Quality control of samples and their packaging was conducted by visual inspection, mechanical, optical, water content, analysis of physical dimensions and collagenase degradation tests on the UVC-exposed samples and non-sterilized samples as controls. Energy intensity of the pulsed UVC light at all positions of the sterilization tray was measured using a UVC Nova II Laser Power/Energy Meter (Ophir Spiricon Europe GmbH) and a LiteMark-XL light intensity monitor.
Optical transparency
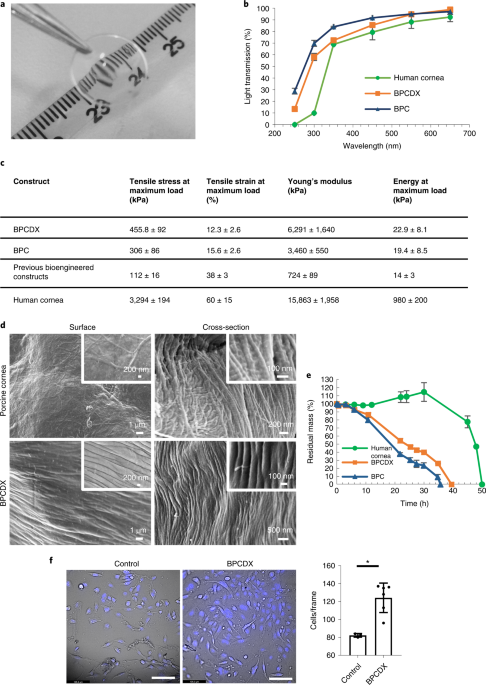
Light transmission through BPCDX was measured across the UV and visible light spectrum (200–700 nm) at room temperature, using a High Performance USB4000 UV-Vis Spectrophotometer (Mettler Toledo). To enable direct comparison with light transmission through the native human cornea, BPCDX samples were 550 µm thick. Samples were immersed in PBS during measurement, and light transmission was compared with published data from healthy human corneal tissue35. Measurements were made for three independent samples per spectrum.
Mechanical properties of scaffolds
Tensile strength, elongation at break (elasticity), elastic modulus (stiffness) and energy at break (toughness) of BPCDX were measured with an Instron Automated Materials Testing System (Model 5943 Single Column Table Frame) equipped with BlueHill software (v.3), a load cell of 50 N capacity and pneumatic metal grips at a crosshead speed of 10 mm min−1. Test specimens were made by molding and curing the samples into dumbbell-shaped Teflon molds followed by equilibration in PBS. Specimens were attached to the grips and tensile force was applied until the sample break point. Data were automatically recorded by the software, and 22 dumbbell-shaped samples were used for each mechanical test.
Characterization by scanning electron microscopy
Morphology of BPCDX and the native porcine cornea was investigated using a Zeiss SEM (LEO 1550 Gemini). PBS-equilibrated samples were washed in water, frozen overnight at −80°C and lyophilized for 12 h. Samples were cut and attached onto metal holders using conductive double-sided tape, and sputter coated with a gold layer for 60 s at 0.1 bar vacuum pressure (Cressington Sputter Coater, 108) before SEM examination. SEM micrographs were taken at various magnifications at 25 kV and 5 kV for porcine cornea and BPCDX samples, respectively.
Enzymatic degradation test
Test samples of BPCDX, a version (BPC) that was singly crosslinked with EDC-NHS29 and donor human cornea, all with a thickness of 500 µm, were exposed to collagenase Type I (from Clostridium histolyticum) and their residual mass percentage as a function of time was measured. Trizma base (Tris base), 2-amino-2-(hydroxymethyl)-1,3-propanediol was used for preparing Tris-HCl buffer. Test samples were equilibrated in 5 ml 0.1 M Tris-HCl buffer (pH 7.4) containing 5 mM CaCl 2 at 37 °C for 1 h. Subsequently, 1 mg ml−1 (288 U ml−1) collagenase solution was added to give a final collagenase concentration of 5 U ml−1 (17 µg ml−1). The solution was replaced every 8 h to retain sufficient collagenase activity. Samples were weighed at various time intervals after gently blotting away the surface water. Three replicates of each sample type were tested. The percent residual mass of the sample was calculated according to the ratio of initial sample weight to the weight at each time point.
In vitro cell biocompatibility
Culture of human corneal epithelial cells (HCE-2 50.B1 cell line, lot no. 70015331, ATCC) was established according to the manufacturer’s instructions. In brief, serum-free keratinocyte growth medium (1×, Gibco) was supplemented l-Glutamine, 5 ng ml−1 epidermal growth factor and 0.05 mg ml−1 bovine pituitary extract (BPE), 500 ng ml−1 hydrocortisone and 0.005 mg ml−1 insulin (Gibco). A T-75 cell culture flask was pre-coated with a mixture of 0.01 mg ml−1 fibronectin, 0.03 mg ml−1 bovine collagen type 1 and 0.01 mg ml−1 bovine serum albumin (BSA), and was incubated overnight at 37 °C. The next day, the excess coating was aspirated, and the flask was allowed to stand for 15 min before seeding the cells. The HCE-2 cell vial was thawed, and cells were seeded at a density of 104 cells per cm2 on the pre-coated flask. Cells were incubated at 37 °C at 5% CO 2 , and growth medium was changed every other day.
BPCDX (300 µm thick) pre-cut to 8 mm diameter were rinsed in PBS and equilibrated in the complete keratinocyte serum-free medium for 2 h in a humidified cell culture incubator. BPCDX samples were then laid down (concave side down) onto the bottom of a 48-well cell culture plate and allowed to adhere to the bottom of the plate for 2 h in an incubator at 37 °C. Three wells were used for BPCDX samples, and three were used as controls (that is, no biomaterial attached to the bottom of the well).
Upon confluence of the seeded HCE-2 cells in the culture flask, the cells were trypsinized with Trypsin-EDTA, then trypsinization was stopped with a complete growth medium and cells were harvested, counted and seeded into the six prepared wells. Cells were seeded at a density of 105 cells per well for the control and BPCDX-covered wells and incubated for 1 h before adding additional medium to reach a final volume of 200 µl growth medium per well. Growth medium was changed every other day, and cells were maintained in culture for 16 days. On day 16, the cells were washed and covered with fresh medium. Cells were stained with NucBlue Live cell stain (Hoechst 33342, Thermo Fisher Scientific) according to the manufacturer’s instructions. Images of stained cells were captured using a Leica DMi8 inverted live-cell microscope under ultraviolet light excitation (385 nm) to detect live cells with fluorescent blue nuclear stain. Brightfield images were additionally obtained to observe cell morphology.
Biological evaluation according to ISO 10993-1:2018
Biocompatibility testing of the BPCDX was performed in conformance to GLPs as per US FDA CFR Title 21 Part 58 and according to ISO 10993 and ISO 11979 standards by an external GLP-certified contract research organization (BioNeeds Private Limited, India). BPCDX underwent the following in vitro and in vivo biocompatibility studies:
1. ISO 10993-3: genotoxicity, carcinogenicity and reproductive toxicity (bacterial reverse mutation tests (ref. report no. BIO-GT-348)) 2. ISO 10993-3: genotoxicity, carcinogenicity and reproductive toxicity (in vitro mammalian chromosome aberration test (ref. report no. BIO-GT-349)) 3. ISO 10993-3: genotoxicity Ames Test (ref. report no. BIO-GT-358) 4. ISO 10993-3: genotoxicity, carcinogenicity and reproductive toxicity (mammalian erythrocyte micronucleus test in Swiss albino mice) (ref. report no. BIO-GT-350) 5. ISO 10993-4: in vitro hemolysis test (ref. report no. BIO-TX-1575) 6. ISO 10993-5: in vitro cytotoxicity: a. In vitro cytotoxicity (direct contact method) (ref. report no. BIO-GT-346) b. In vitro cytotoxicity (elution method) (ref. report no. BIO-GT-347) 7. ISO 10993-10: skin sensitization in guinea pigs (ref. report no. BIO-TX-1574) 8. ISO 10993-10: in vivo ocular irritation in rabbits (polar + non-polar) (ref. report no. BIO-TX 1584) 9. ISO 10993-11: acute systemic toxicity in Swiss albino mice (ref. report no. BIO-TX-1576)
Bacterial endotoxin test according to ISO 11979-08
To ensure the sterile BPCDX would be safe for human use, endotoxin testing was performed internally and by an independent laboratory (S2 Medical AB) using an Endosafe-PTS endotoxin analyzer (Charles River) with a rapid, point-of-use spectrophotometer and USP/BET-compliant disposable cartridges for real-time endotoxin testing. The limulus amebocyte lysate cartridges used in the tests are FDA-licensed for in-process and final product release testing, ensuring regulatory compliance of the output results. In brief, the tests were performed and reported according to ISO 11979-8 (ophthalmic implants-intraocular lenses-part 8: fundamental requirements amendment 1) and ISO 15798. The acceptable endotoxin limit for ophthalmic devices as specified in ISO 15798 is 0.2 EU per device against which BPCDX test results were evaluated.
Shelf-life stability tests according to ISO 11607
Verification of stability is a time-consuming and resource-intensive task in the development of new medical devices and is often overlooked in research studies. For wide distribution of the BPCDX to regions with the greatest unmet need, shelf-life studies are critical to ensure the device produced in a normal manufacturing process functions as intended despite logistical, storage and other barriers that may occur in the distribution chain. Often devices are tested under accelerated conditions to increase the rate of chemical and/or physical degradation and therefore decrease the time required to obtain stability data. Long-term or real-time studies are still needed however, as the accelerated and long-term results may differ. For packaged and sterilized BPCDX, we therefore performed an accelerated shelf-life stability study by incubating devices at 28 °C for 6 months, and we performed a real-time shelf-life stability study by incubating devices at 7 °C for 2 years. Control BPCDX samples not subjected to aging (time-zero samples) were prepared and tested for visual appearance, mechanical properties, optical transmission, water content, size, collagenase degradation, in-house tryptic soy broth sterility test and for sterility tests conducted by an independent GMP-certified laboratory (MIKROLAB Stockholm AB). The accelerated and real-time aged samples were also tested for the above properties and compared with the control samples.
In vivo biocompatibility, subcutaneous implantation in rats
To test compatibility of BPCDX after surgical implantation in vivo, we used a model of subcutaneous implantation in rats as described previously43. Three male Wistar rats aged 8 weeks were given a pre-operative analgesic (0.01 mg buprenorphine) by intraperitoneal injection the day before surgery, day of surgery, and 1 and 2 days after surgery. Under general anesthesia (25 mg ml−1 ketamine and 0.5 mg ml−1 dexmedetomidine hydrochloride), a 2-cm long paravertebral cutaneous incision was made into the dorsal flank of the rat, after which a subcutaneous pocket was created by blunt dissection. A 1-cm square piece of the BPCDX was inserted into the pocket, and the pocket was sealed with three absorbable 9-0 Vicryl sutures. Eight weeks post-implantation, rats were euthanized, and the tissue region surrounding the implant zone was excised and prepared for immunohistochemical analysis. The procedures were performed after obtaining ethical approval from the Linköping Animal Ethical Review Board (permit ID 585), with procedures in compliance with EU Directive 2010/63/EU on the protection of animals used for scientific purposes.
Minimally invasive BPCDX implantation in minipigs
To evaluate implantation of BPCDX in vivo in the cornea, we designed a model of keratoconus in the Göttingen minipig to create an artificially thin native corneal stroma mimicking the pathologic thinning in advanced keratoconus. To achieve this, we used an ophthalmic femtosecond laser (iFS 150, Abbott Medical Optics) and modified the technique we previously reported in rabbit models, termed FLISK29,31. Animal experiments were performed after receiving approval from the Linköping Animal Ethical Review Board (permit ID numbers 153 and 37–16) and adhered to the guidelines of the ARVO Statement for the Use of Animals in Ophthalmic and Visual Research and the Directive 2010/63/EU. All surgeries were performed in a licensed large-animal surgical suite with controlled temperature and humidity at the Linköping University Translational Medicine Center, under supervision of the university veterinarian and animal care team. Ten female Göttingen minipigs (Ellegaard Göttingen Minipigs A/S) aged 6 months and weighing 15 kg were pre-medicated with sedatives (3 mg kg−1 tiletamine HCl + 3 mg kg−1 zolazepam HCl, Zoletil and 0.06 mg kg−1 medetomidine HCl, Dexdomitor), administered intramuscularly. Anesthesia was initiated with 0.2 mg kg−1 Propofol, administered intravenously through a venous catheter placed in the ear vein. Thereafter, the animals were intubated with anesthesia maintained using 0.5–3.0% isoflurane gas. During anesthesia, hydration was maintained by intravenous administration of Ringer’s acetate solution at 10 ml kg−1 h−1. Immediately before surgery 0.02 mg kg−1 atropine was given by intramuscular injection. Topical anesthesia (0.5% tetracaine HCl eye drops) was given before laser surgery, and all surgeries were performed in a single eye per animal.
To achieve a thinner stroma mimicking advanced keratoconus, we used the femtosecond laser to cut a central mid-stromal button in the cornea 7 mm in diameter and 250 µm in thickness, with the anterior surface of the button located 200 µm below the corneal surface (Supplementary Fig. 2). To achieve this we pre-programmed the laser to perform cuts in the following order: a posterior circular lamellar planar cut of 7.1 mm diameter located 450 µm below the corneal surface, a 360° circular side cut of 7.0 mm diameter extending from the posterior lamellar plane 250 µm anteriorly, and an anterior circular lamellar plane of 7.1 mm diameter at a depth of 200 µm below the corneal surface, followed by a final 90° arc-shaped access cut, oriented at 45° to the lamellar planes and extending to the epithelial surface. Details of the femtosecond laser protocols are given elsewhere65. Following the laser procedure, we used a blunt hockey blade tool to separate the access cut and lamellar planes, and surgical forceps to extract the button of native stromal tissue through the access cut. This resulted in a cornea approximately two-thirds the thickness of the normal porcine cornea, mimicking the cornea in advanced keratoconus, albeit with a uniform reduction in thickness across the central 7 mm of cornea.
We thereafter conducted minimally invasive surgery to treat advanced keratoconus. A sterile, packaged BPCDX implant of 280 µm thickness and 10 mm diameter was opened in the operating room, and we cut the device to a 7 mm diameter using a Barron corneal punch trephine. Next, we used surgical forceps to grip the BPCDX, which was then inserted into the intrastromal pocket through the access cut65. Although the FLISK surgery was previously performed without the use of surgical sutures29, as a precautionary measure we decided to close the region of the access cut in the stroma anterior to the implant using two 10-0 nylon surgical sutures (Supplementary Fig. 2).
Immediately post-operatively, minipigs were placed on a ventilator and once spontaneous breathing resumed, 0.05 mg kg−1 intravenous buprenorphine (Temgesic) was given as an analgesic. Post-operatively, topical anesthetic eye drops were again instilled, followed by a combination topical corticosteroid-antibiotic (0.1% dexamethasone + 0.3% tobramycin, Tobrasone eye drops) given three times daily the first post-operative week and twice daily during the following three weeks. Post-operative analgesia consisted of intramuscular injection of 0.05 mg kg−1 buprenorphine every 12 h for the first five post-operative days.
Post-operative assessment and corneal imaging
Six months after surgeries, minipigs were placed under general anesthesia as described above, and we performed in vivo examinations and photo documentation in operated eyes. Examinations consisted of digital photography (Nikon D90 digital camera), anterior segment optical coherence tomography (iVue, Optovue) and in vivo confocal microscopy (Heidelberg Retinal Tomograph 3 with Rostock Corneal Module, Heidelberg Engineering) using a previously described microscopy protocol66. Following examinations and data collection, minipigs were euthanized while under general anesthesia and deep sedation, by intramuscular injection of 7 mg kg−1 Zoletil and intravenous injection of 100 mg ml−1 pentobarbital.
Histology and Immunohistochemistry
Following euthanasia, rat tissue and porcine corneas (to the limbal margin) were dissected under an operating microscope, embedded in optimal cutting temperature compound, and snap-frozen in liquid nitrogen. Corneas were stored at −80 °C until further use. For histology, rat and pig tissue were thawed, fixed in 4% paraformaldehyde solution, embedded in paraffin and sectioned to a thickness of 4 µm, followed by staining with hematoxylin and eosin (H&E). For immunohistochemistry, sections 10 µm thick were produced using Leica CM1510 cryostat (Leica AB). The resulting sections were mildly fixed with 2% paraformaldehyde (VWR Life Science) for 10 min, permeabilized by incubation in 0.05% Triton X-100 for 10 min and blocked with 5% BSA for 1 h at room temperature. The samples were then incubated overnight at 4 °C with primary antibodies β-III-tubulin (1:100, ab7751, clone TU-20, lot GR3238448-11, Abcam), α-SMA (1:25, ab7817, clone 1A4, lot GR119216-7, Abcam), type III collagen (1:100, Acris AF5810, clone III-53, lot A130097BH) and CD45 (1:100, ab23, clone UCH-L1, lot GR3189280-2, Abcam) in 2.5% BSA. Control sections were incubated with 2.5% BSA alone without the addition of the primary antibody. Sections were washed in PBS-T and visualized by goat anti-mouse IgG (H + L) Alexa Fluor 488 (1:1,000, A32723, polyclonal, RRID AB_2633275, Thermo Fisher Scientific) or DyLight 488 (1:1,000, 35503, polyclonal, RRID AB_844397, Thermo Fisher Scientific) and DyLight 550 (1:1,000, SA5-10173, polyclonal, RRID AB_2556753, Thermo Fisher Scientific) secondary antibodies. Cell nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI); (1:5,000; Sigma). The slides were mounted with a ProLong Diamond antifade reagent (Invitrogen, Thermo Fisher) and imaged with a laser fluorescence confocal microscope (LSM700, Zeiss).
Ethics statement
BPCDX was implanted intrastromally in human subjects in local investigator-driven pilot feasibility studies in Iran and India. BPCDX was implanted in cases of advanced keratoconus causing severe visual impairment or corneal blindness. Before subject recruitment, ethical approvals for the studies were obtained in Iran (Institutional Review Board, Farabi Hospital, Tehran University of Medical Sciences, Tehran, Ethics Approval Code IR.TUMS.FARABIH.REC.1395.442) and in India (Institute Ethics Committee, All India Institute of Medical Sciences, New Delhi, ref. no. IEC/NP-47/10.04.2015, RP-23/2015). Studies were conducted following the tenets of the Declaration of Helsinki, and signed informed consent was obtained from all participants before inclusion. LinkoCare Life Sciences AB sponsored the pilot studies, which were funded by LinkoCare Life Sciences AB in kind (both sites), Care Group India (Indian study) and the local investigators in India and Iran. No European Union funding was used for clinical activities outside of the EU. The study was an exploratory, non-randomized, non-blinded and non-controlled pilot case series whose primary goal was to test feasibility of a new surgical method and detect possible adverse reactions in the host or in the BPCDX device using different implanted device thicknesses, analogous to an initial dosing study to determine drug tolerance. The rationale for conducting the pilot study was to obtain first feasibility and safety/tolerability data to determine if it would be ethically acceptable to randomize visually impaired patients (a vulnerable group) to possibly receive the experimental treatment and not a standard donor cornea and surgery when available, as would be required for a future randomized controlled trial. Broad dissemination of pilot safety and feasibility data was not initially foreseen, and local investigators initiated surgeries immediately upon receiving local ethical approvals, without pre-registration of the pilot series in a public registry. Interim analysis of pilot data indicated safety and revealed an unexpected efficacy comparable to outcomes of standard transplantation surgery (DALK or PK) with possible additional clinical benefits from the milder surgery. On the basis of the interim data, approvals for randomized controlled studies were granted in the EU (see below). For these reasons, publication of initial pilot feasibility results including details of the proposed minimally invasive surgery would be in the interest of the medical and scientific community, before initiating a randomized controlled trial. To ensure the reporting of the pilot studies in Iran and India would be consistent with best practices for the conduct of investigational studies of medical devices, the ongoing pilot clinical study was registered in the ClinicalTrials.gov database in December 2020 (Clinicaltrials.gov: NCT04653922). The BPCDX manufacturing process and device test results presented here, along with preclinical data and pilot clinical results at 6–12 months of follow-up, were submitted to Regulatory Authorities in Sweden (Medical Products Agency) and the Czech Republic (State Institute for Drug Control), and approvals were granted for a randomized controlled study protocol according to EU Directive 93/42/EEC on Medical Devices (decision 5.1-2018-44565, Sweden; file no. sukls 21920/2020, Czech Republic) and for the BPCDX device manufacturing, sterilization and packaging methods (file no. sukls 21920/2020, Czech Republic). Additionally, an institutional ethical review committee in Sweden reviewed a randomized clinical trial protocol and approved it for use in Sweden (Linköping Regional Ethics Committee, decision 2017/34-31). Because BPCDX device development and preclinical studies were partially funded by the European Union (Project ARREST BLINDNESS, grant no. 667400), before any reporting of results the clinical studies conducted in Iran and India were subject to further review by an independent contracted biomedical ethics expert (H. Draper, University of Warwick, UK) and by an independent Ethics Review Panel at the European Commission, Division of the Director General—Research and Innovation. The outcome of these reviews was favorable, confirming that the conduct of the clinical studies was consistent with accepted ethical practices within the EU and that the studies were conducted without exploitation of the research subjects or local investigators.
Pilot feasibility study according to ISO 14155
To investigate the safety and feasibility of BPCDX use in LMICs, we conducted pilot studies in Iran (Farabi Eye Hospital, Tehran University of Medical Sciences, Tehran, Iran) and India (All India Institute of Medical Sciences, Dr. R. P. Centre for Ophthalmic Sciences, New Delhi, India). The aim of these pilot studies was to set broad guidelines for the use of the BPCDX device (such as inclusion and exclusion criteria and parameters for intrastromal surgery) to determine the feasibility to implement the proposed treatment in local patient populations, while allowing surgeons the flexibility to adapt to local clinical procedures, protocols and surgeon experience and preferences. Several parameters therefore varied between sites such as the size of BPCDX and size of intrastromal pocket, choice of post-operative medications and timing and conduct of follow-up examinations using instruments and diagnostic methods available to investigators at the local clinics. The goal of these initial case series was to obtain initial safety, feasibility and efficacy data, which if successful, would support the design and implementation of future prospective, randomized, controlled clinical trials.
Ethical permission was granted for the pilot studies to treat up to 20 subjects with advanced keratoconus at each site (40 subjects total), based on the ability to detect adverse events in 10% of cases. We report results of 24-month follow-up of the first 12 patients treated in Iran and the first 8 patients treated in India (20 patients total). For the results reported here, clinical data collection occurred during February 2017–January 2020 in Iran and during November 2016–March 2020 in India. In addition to safety and feasibility of BPCDX implantation in advanced keratoconus using the minimally invasive FLISK procedure, the study sites collected data to enable assessment of possible efficacy in terms of rehabilitation of corneal curvature, corneal thickness and BCVA. A future definitive trial would be based upon occurrence of adverse events (for example, inflammation and rejection) leading to implant removal in a maximum of 10% of cases, along with evidence of efficacy in the form of at least 60% of operated eyes having sustained decrease of keratometry at 6 months, sustained increase in central corneal thickness at 6 months, and minimum visual acuity improvement of one Snellen line of vision at 6 months.
Subject recruitment and study endpoints
Potential study subjects at both sites were identified on the basis of clinical history, previous consultation visits and fulfillment of study inclusion/exclusion criteria. Potential candidates were contacted by telephone by the study nurses, then sent the study information and consent form by postal mail. If a subject decided to participate in the study, the consent form was signed by the subject and the local investigator-physician in charge of the study, and the subject was formally included. Study participants were not compensated for their participation in the study, monetarily or otherwise. The primary endpoint for this study was to determine the safety and stability of BPCDX implantation, defined as retention of the device without degradation, loss of transparency, inflammation or vascularization of the implant or host cornea during the first 6 post-operative months. Primary outcome measures included safety (determined by maintenance of corneal transparency and absence of rejection, scarring, inflammation or neovascularization detected during clinical examinations up to 6 months post-operatively) and efficacy measures to assess reduction in maximum keratometric power, increase in corneal thickness and improvement in uncorrected and BCVA at 6 months. The secondary endpoint was to determine safety during a 12-month post-operative period, while secondary outcomes were the same safety and efficacy measures as above, but at 12 months. Here we report the longer-term 24-month results for the primary study endpoint in the first 20 operated subjects.
Inclusion and exclusion criteria for subject selection
Subjects were eligible for study inclusion if the following criteria were all met in at least one eye:
1. Grade 3 or higher advanced keratoconus (according to Amsler–Krumeich classification). 2. No corneal scar. 3. Male or female aged ≥18 years. 4. Subjects indicated for a first corneal transplantation. 5. Corneal thickness (including epithelium) at least 300 µm centrally, as measured by OCT. 6. Signed and dated informed consent.
Patients fulfilling at the selection visit one or more of the following exclusion criteria were not included in the study:
1. Ophthalmic exclusion criteria In the affected eye: Prior corneal surgery (for example, refractive surgery, cataract, collagen crosslinking, endothelial keratoplasty, etc.). In either eye:
Dry eye/tear film pathology.
Active ocular infection.
Glaucoma/ocular hypertension.
Active corneal ulceration.
Acute or chronic disease or illness that would increase the operation risk or confound the outcomes of the study (immune-compromised, connective tissue disease, clinically significant atopic disease etc.).
Any other medical condition that in the judgment of the local clinical investigator was not compatible with the study procedures. 2. Systemic/non-ophthalmic exclusion criteria General history judged by the investigator to be incompatible with the study (for example, life-threatening patient condition or other condition where post-operative follow-up may be difficult).
Known diabetes or other neuro-degenerative disorder (as corneal nerves can be affected leading to impaired wound healing). 3. Exclusion criteria related to general conditions Inability of patient to understand the study procedures and thus inability to give informed consent.
Participation in another clinical study within the last 3 months.
Already included once in this study (can only be included for one eye).
Surgical method for clinical case series
We used the minimally invasive FLISK method at both sites. All study subjects were candidates for penetrating or lamellar keratoplasty. The diagnosis of advanced keratoconus was made according to clinical signs (Munson’s sign, Rizutti’s sign) slit-lamp biomicroscopic examination (Vogt’s striae, Fleischer’s ring, apical thinning), corneal topography (skewed asymmetric bow-tie, severe central or inferotemporal steepening, high keratometric power), tomographic signs (abnormal elevation maps, abnormal pachymetry maps, keratoconus detection by Belin/Ambrosio Enhanced Ectasia Display) and refractive results (loss of BCVA). Subjects with refractive errors that could not be corrected with eyeglasses or routine contact lenses, and those who were scleral or mini-scleral contact lens intolerant were included. After the corneal center was marked on the basis of the pupil center, a mid-stromal pocket was created through the marked margins of the temporal cornea using a femtosecond laser. The laser surgical parameters are summarized in Supplementary Fig. 3. Following the laser procedure, the corneal stroma was dissected, taking care to avoid perforation of the Descemet membrane while dissecting the thinnest part of the cornea. Next, BPCDX was removed from the sterile packaging and trephined at 7.5–8.5 mm size and inserted into the intrastromal pocket using surgical forceps to grip the BPCDX across its entire diameter. In Iran, two different thicknesses of BPCDX were used, 280 µm and 440 µm, on the basis of the pre-operative corneal thickness at the thinnest point. Subjects with a thinnest value above 400 μm received 280-μm-thick BPCDX while those with thinnest value below 400 µm received 440-μm-thick BPCDX. In India, all subjects received 280-µm-thick BPCDX regardless of initial corneal thickness. No suturing was performed in any subject. Immediately post-operatively, a soft silicone hydrogel bandage contact lens (Comfilcon A, CooperVision) was placed on the operated cornea for 3 days in all treated subjects.
Post-operative medications and follow-up examinations
Post-operatively, medications were administered as deemed appropriate by the local surgeons. On the basis of the post-operative wound healing observed in the minipig model (where post-operative medications were administered for 4 weeks) and in a previous rabbit study where the intrastromal implantation model indicated complete wound healing within 8 weeks29, an 8-week regimen was instituted. In Iran, patients received artificial tears (carboxymethylcellulose 0.5%) as needed pre-operatively and three times daily post-operatively for 8 weeks, and betamethasone eye drops (0.1%) were given three times daily for 8 weeks post-operatively. Additionally, a bandage contact lens was placed over the operated cornea for 1 week. In India, patients received pre-operative moxifloxacin eye drops (0.5%) three times daily for 3 days prior to surgery. Post-operatively, patients received moxifloxacin eye drops (0.5%) three times daily, prednisolone eye drops (1%) four times daily and artificial tears (carboxymethylcellulose 0.5%) six times daily.
Surgeons assessed the eye immediately following surgery and at 1 day, 1 week and 1, 3, 6 and 12 months post-operatively. An additional 24-month visit was subsequently scheduled. Slit-lamp biomicroscopic evaluation was used to grade the corneal transparency according to a previously published scale67, uncorrected visual acuity and BCVA were determined by the Snellen eye chart and spectacles, and expressed in the logMAR scale, Scheimpflug-based anterior segment tomography was performed to assess corneal steepness (Pentacam HR, Oculus, Optikgerate GmbH), and refraction was assessed at various post-operative visits. Corneal thickness was assessed using anterior segment FD-OCT (Iran: Casia, Tomey; India: iVue, OptoVue). Pre-operative and post-operative corneal imaging examinations were performed by the same experienced optometrist at each site. Data was entered into a spreadsheet for later analysis (Microsoft Excel 365 for Windows, 32-bit).
Statistical analysis
Statistical analysis was performed using Statistical Package for Social Sciences software (v.22, SPSS). Normality in the distribution of the parameters was assessed using the Shapiro–Wilk test. For shelf-life studies, t-tests were performed comparing aged to non-aged samples. To examine differences in post-operative values for clinical parameters at 24 months relative to the pre-operative values, a two-tailed paired t-test was performed. Where results across two different groups were compared, an independent t-test was performed. A two-tailed critical value of α < 0.05 was considered statistically significant for all tests, unless otherwise stated. Visual acuity comparisons were made in logMAR units, with each line of improvement in acuity corresponding to a logMAR reduction of 0.1.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.